今日,顶尖学术期刊《自然》在线发表了一篇关于基因编辑的重要论文。来自Broad研究所的CRISPR大牛刘如谦(David Liu)教授团队开发了一种新型的基因编辑技术,有望修复大约89%的已知人类致病变异。这项研究在发表之前,就已经在一些学术会议上得到了很多科学家的关注。正式上线后,更是引起了业内的点赞和热议,被认为是“极为重要”的成果。
那么,这项技术究竟牛在哪里?要讲清楚这一点,我们还要先看看传统的基因编辑是怎么做的。
目前,应用最为广泛的基因编辑技术之一,便是CRISPR-Cas9系统。在这套系统里,Cas9蛋白会在目标DNA序列上切出口子,造成双链DNA断裂。随后,再利用同源重组等方法,在DNA修复的过程中对基因组进行编辑。
这一方法虽然有效,但很粗暴。打个比方,这就好像一页书上有错别字,为了修正书上的错误,要把这整页书给撕下来,再重新粘上一页那样。可以想象,这样的工作谈不上优雅,也容易产生潜在的问题。
过去,包括刘如谦教授课题组在内,一些科学家们开发出了“单碱基编辑系统”。与传统的CRISPR-Cas9系统不同,它将经过修饰的Cas9蛋白与另一种酶直接融合在一起。在目标DNA位点,它不会切开DNA链,而是对单个碱基进行修改,如将A变成G,或是把C变成T。这一方法能够对点突变进行“微调”,不过并不适合大段大段的修改。此外,它只能进行四种修改(比如不能把T变成A),还存在潜在的脱靶问题。
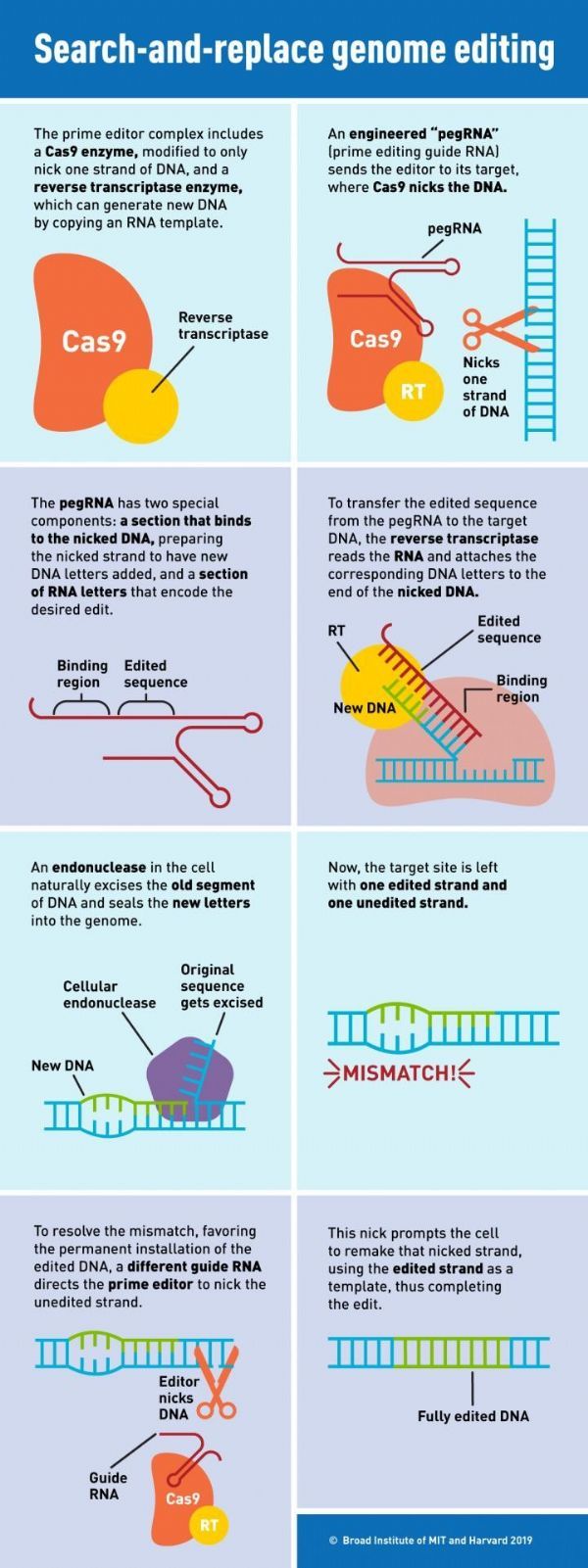
而在今日的这项研究中,我们见证了一种新型基因编辑技术的诞生。这种技术被称为“先导编辑”(prime editing),核心之一是一种由Cas9蛋白与逆转录酶(reverse transcriptase)融合而成的特殊蛋白。该技术的另一个关键是一种特殊的gRNA,它被研究人员们称为“pegRNA”,其中pe是“先导编辑”英文单词的两个首字母。
与普通的gRNA不同,这种pegRNA不但能够结合想要进行编辑的特定DNA区域,还自带“修改模板”。Cas9-逆转录酶融合蛋白会在pegRNA的引导下,精准地切开一条DNA链,然后根据“修改模板”,合成含有正确序列的DNA。细胞内的DNA修复机制会自动把这段新合成的序列整合进基因组。
随后,研究人员们故技重施,在另一条DNA链上进行同样的操作。这样一来,两条DNA都能得到正确的编辑。
“结果就是(给基因组带来)永久的改变,这些改变来源于pegRNA所编码的信息。”刘如谦教授说道。
正是由于这套系统的灵活性,原本不可能实现的单碱基编辑(如将T转变为A),也成为了可能。与单碱基编辑只能进行4种转换相比,这套先导编辑系统能进行12种单碱基的转化。也就是说,他们可以把任何碱基转化为任何其他碱基,包含了所有的12种可能性。
在多种人类细胞中,研究人员们证实了这一系统的编辑能力。此外,它的编辑潜力在小鼠神经元里也得到了证实。除了对单个碱基进行修改之外,这一系统还能够插入或删除特定的DNA序列。根据论文,研究人员们最多可以插入44个碱基,或是删除80个碱基。
“通过先导编辑技术,我们能直接修正镰状细胞贫血症的突变(学术经纬注:A突变成了T),让序列回归正常。我们也能移除戴萨克斯症(Tay Sachs disease)多出的4个碱基。这都不需要完全切开DNA,或添加DNA模板,”刘如谦教授在Broad研究所的新闻稿里说道:“这一系统的美丽之处在于几乎对编辑的序列没有限制。由于添加上的碱基仅由pegRNA来决定,我们能加上与原本序列仅差1个碱基的序列,也可以加上多了几个,或少了几个碱基的序列。我们还可以对这些变化进行排列组合。”
可喜的是,研究人员们还发现,与需要断开双链DNA的传统方式相比,先导编辑系统的脱靶编辑风险更低。
目前,这一系统已向学术界和非营利组织免费公开。
当然,由于这一系统依旧是一类全新的基因组编辑方法,因此还需要进一步的检验和改良,才能用于人类。不过,这一系统展示的潜力,已经让我们看到了一个能够有效治疗罕见遗传病的未来。我们期待这一天的早日到来。(生物谷Bioon.com)
 基因君官网
基因君官网