 基因君官网
基因君官网
国立首尔大学医学院的研究人员和美国被誉为“测序黑马”的公司——10x Genomics联合运用去年新发售的GemCodeTM测序平台以及第三代测序PacBio单分子实时测序平台在本期的Nature上发表了一项新的研究,对一名韩国人的基因组(AK1)进行从头组装和单倍体型定相信息分析。这是迄今为止发表的最为连续的人类基因组组装。作者填补了特异人群参考基因组的空白,并确定了结构变异。
该研究的显着改进
这项工作提供了了到目前为止最邻近的二倍体基因组装配与之前未报道的亚洲特异结构变异的广泛调查。其优质的临床相关的等位基因的单体型分析有着提高精准医疗的潜力。这对于目前基于欧洲人和白人人群的基因组分析方法,是一个显着的改进。这个方法提高了从一个个体的短读长序列比对形成到一个单倍体的一致性参考基因组的分析状况,调整了通常无法捕捉到相对于个体基因组或种族人群属于新序列的问题。
主要研究方法
GemCode平台能够与现有的短读取测序仪互补,产生长片段信息(10-100 kb),实现结构变异和单体型等分析。为了更全面了解结构变异的范围和更好地理解AK1二倍体基因组结构的相,研究者对从PacBio单分子实时测序reads组装得到的,每个染色体单倍型的装配体——“haplotig”进行从头组装。除了PacBio长读长测序,该研究还使用了Illumina短读长测序、10X Genomics连接读取、细菌人工染色体(BAC)测序和BioNano Genomics光学图谱。Contig N50达到17.9Mb,Scaffold N50达到44.8Mb。
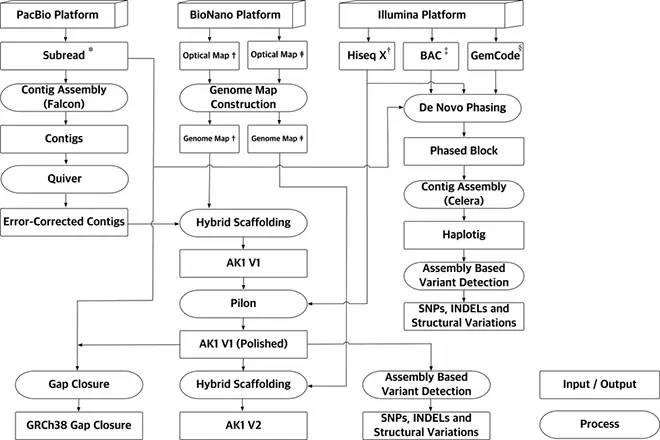
实验设计及数据分析流程
数据上的提升
到目前为止该研究在之前最佳的N50长度上提高了18Mb,最大的91个scaffold覆盖了基因组的90%,8个染色体的臂是由单个scaffold构成的。研究人员用AK1的组装序列补齐了在人类参考基因组GRCh38上的190个空缺中的105个,补齐的序列由364kb变成1.5Mb。另外也将剩下的85个空缺中的72个从663kb补充到4.1Mb。该研究鉴定到的18,210个结构变异,包含7,358个删除,10,077个插入,71个转换和704个复杂变异,所有这些变异均在AK1组装结果和人类参考基因组GRCh37之间进行了直接比较,达到了单碱基的分辨率。在18,210个结构变异中,总共有11,927个变异是以前未曾报道的。
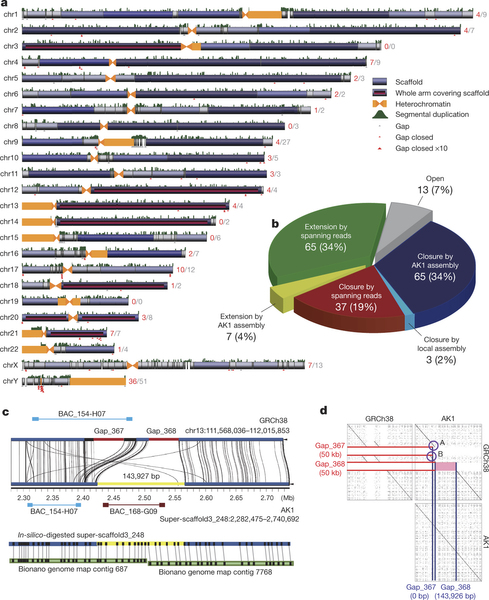
AK1 de novo assembly Scaffolds与GRCh38的数据结果进行比较
测序新技术的强大作用
这项研究的结果显示了测序新技术在从头基因组组装和“真正的”二倍体定相方面的强大能力,可以用来检测全方位的遗传变异和了解临床相关基因的单倍体的结构。这项基因包括:主要组织相容性复合体(MHC)的人类白细胞抗原(HLA)的基因变异,对药物基因组非常重要的CYP2D6基因,以及罕见的常染色体隐性遗传疾病,如家族性地中海热、Upshaw–Shalman综合征中涉及到的基因。
该项研究的领导者,国立首尔大学医学院的Jeong Sun Seo医生说:“10x Genomics的GemCode平台和有针对性的BAC克隆测序在这项研究中成功单倍体定相是必不可少的。”
10x Genomics的联合创始人和首席执行官SergeSaxonov说:“从头装配和定相就像十字瞄准线一样是我们的主要目标。我们的技术非常适合于临床和人群规模的测序。”
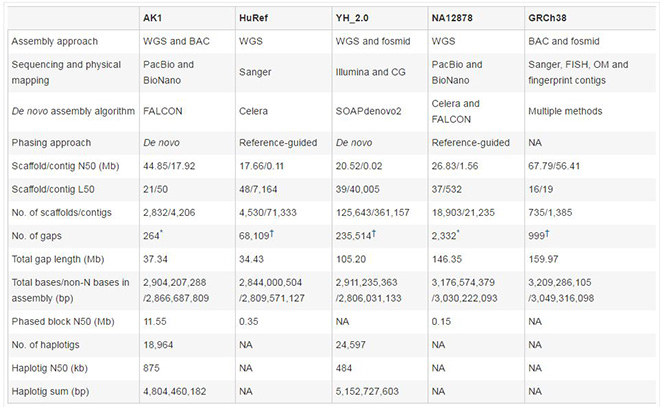
人类基因组从头组装及单倍体型定相信息总结统计
作者在文章中还比较了目前已有的人类基因组组装的测序平台、算法、组装和定相统计数据。该列表表明,单分子长读长测序技术在生成高质量组装数据上远超过短读长测序技术。这项研究清楚地表明了第三代测序长读长技术结合GemCode测序平台解决单倍型的从头组装方法的潜力,并揭示了以前未被发现的新的遗传变异,将迎来人类基因组测序的新标准。(生物谷Bioon.com)
相关会议推荐

2016(第三届)下一代测序发展论坛
会议时间:2016.11.17-2016.11.18 会议地点:上海