 基因君官网
基因君官网

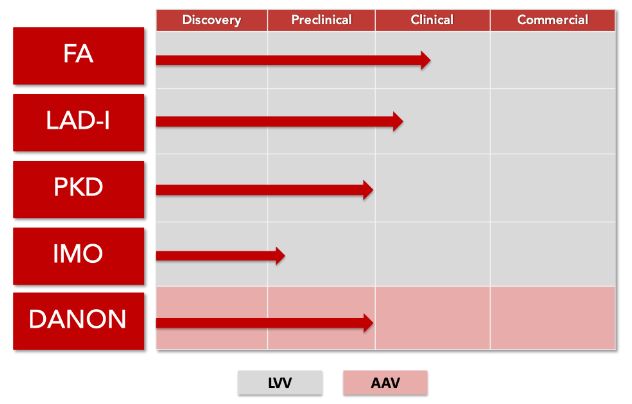
2018年11月29日/生物谷BIOON/–Rocket制药公司是一家新兴的临床阶段生物技术公司,专注于开发一流的基因疗法治疗罕见的、毁灭性的疾病,该公司的多平台开发方法应用了成熟的慢病毒载体(LVV)和腺相关病毒载体(AAV)基因治疗平台。
近日,该公司宣布,美国食品和药物管理局(FDA)已授予其先导项目为RP-L102再生医学先进疗法资格(RMAT)和快速通道资格(Fast Track)。RP-102是一种基于LVV的基因疗法,开发用于范可尼贫血(Fanconi anemia,FA)的治疗。此次RMAT资格的授予,是基于正在欧洲开展的一项I/II期临床研究的积极疗效和安全性结果。
RP-102的治疗流程先是在体外采用CD33+筛选程序去除和分离造血干细胞,用RP-102进行基因修饰,再将自体基因修饰的富含CD34+的造血细胞回输至患者体内以恢复功能。
目前,RP-102治疗FA的研究性新药申请(IND)已获FDA受理。之前,RP-102也已被FDA和欧洲药品管理局(EMA)授予治疗A型FA的孤儿药资格。除了RP-102之外,Rocket公司也正在开发治疗丙酮酸激酶缺乏症(PKD)、白细胞黏附缺陷症I型(LAD-I)、婴儿恶性骨质疏松症(IMO)的基因疗法。
RMAT是2016年12月美国修改“21世纪治愈法案(21st Century Cures Act)”的再生医疗条款时,为了加速创新再生疗法的开发和审批而制定的一种Fast track制度。RMAT可以是细胞疗法、治疗性组织工程产品、人类细胞及组织制品,或是其他包含了再生医学技术制品的联合疗法。在研药物要获得RMAT资格认定,必须要有初步的临床研究数据证明药物在治疗、延缓、逆转或治愈严重或危及生命的疾病或是在未满足医疗需求方具有积极的结果。RMAT资格认定提供了类似于突破性药物资格(BTD)的优惠政策,包括与FDA进行早期互动、优先审查、加速审批的可能性。
FDA的快速通道项目旨在加速针对严重疾病的药物开发和快速审查,以解决关键领域严重未获满足的医疗需求。实验性药物获得快速通道地位,意味着药企在研发阶段可以与FDA进行更频繁的互动,在提交新药申请(NDA)后也可能会获得FDA的优先审查。